What is HAE?
Hereditary angioedema (HAE) is a rare and potentially life-threatening genetic disease affecting ~1:50,000 people. Individuals with HAE experience recurring and unpredictable cutaneous and submucosal oedema of various body sites.1, 2
Hereditary angioedema (HAE) is a rare and potentially life-threatening genetic disease affecting ~1:50,000 people. Individuals with HAE experience recurring and unpredictable cutaneous and submucosal oedema of various body sites.1, 2
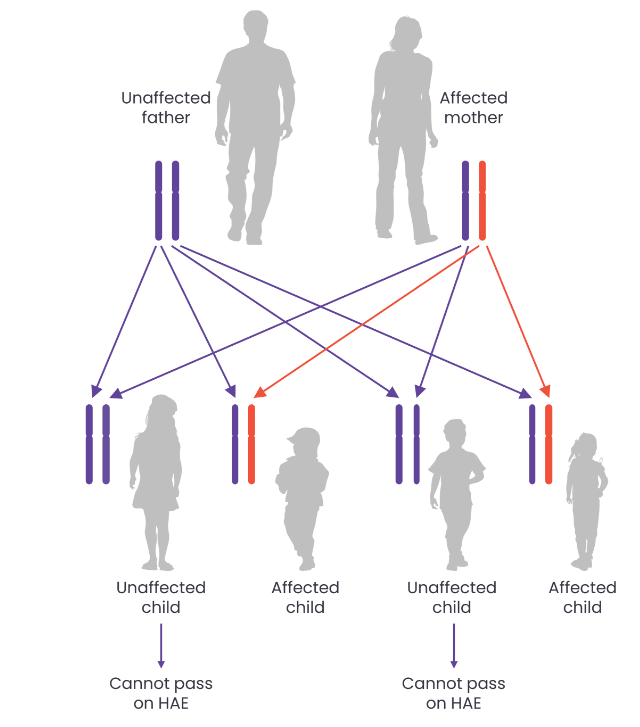
Most cases of HAE are caused by a mutation in the SERPING1 gene which encodes the protein C1 esterase inhibitor (C1-INH), resulting in either a deficiency (type I) or dysfunction (type II) of C1-INH. HAE is an autosomal dominant disease; therefore, it is recommend that family members and offspring of diagnosed patients are also tested for HAE. However, 25% of cases occur due to de novo mutations.1, 3
One of the main roles of C1-INH is to regulate the activity of key proteases of the contact system and therefore regulate bradykinin production. Bradykinin is the key mediator of increased vascular permeability in HAE, which is generated when plasma kallikrein cleaves high molecular weight kininogen. With low functional levels of C1-INH in patients with HAE, bradykinin production is not properly regulated and can be overproduced. During HAE attacks, increased activation of bradykinin B2 receptors in blood vessel walls leads to increased vascular permeability and angioedema of cutaneous and submucosal tissue. The contact system is initiated when factor XII (FXII) is activated and in turn activates plasma kallikrein, which cleaves high molecular weight kininogen to produce bradykinin.1
Watch the video below to learn more about the contact system and the cascade leading to HAE attacks

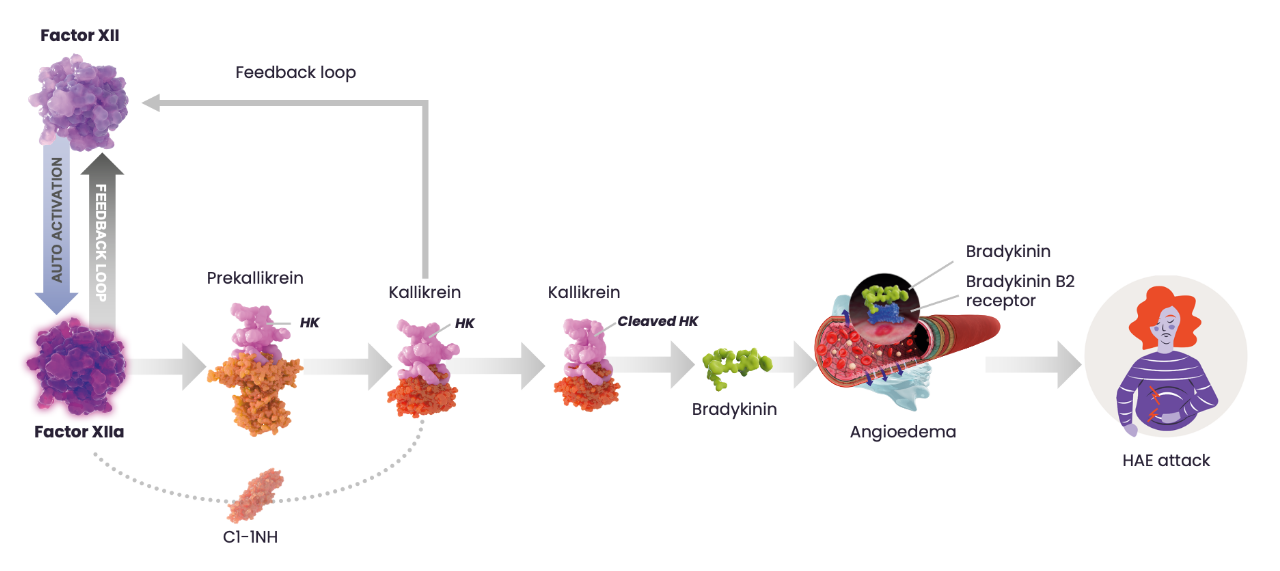
Simplified illustration of the contact activation pathway and its role in an HAE attack. Adapted from Zuraw BL N Eng J Med. 2008. C1-INH, C1-esterase inhibitor; HK, high-molecular weight kininogen. 4-6
Patients usually experience their first symptoms of HAE (HAE attacks) during childhood and adolescence.3 Attacks can occur anywhere on the body but often affect the skin, abdomen and upper respiratory tract. Attacks can be extremely painful, disfiguring and debilitating, interfering with patients’ ability to carry out normal daily activities.2 Symptoms often get worse over a period of 12 hours to a day, then gradually resolve over the following 2–5 days.2

HAE varies greatly from person to person, with some patients suffering several attacks per week, while attacks for others are rare.2 HAE implies a high disease burden that includes the frequency and severity of attacks, as well as detriments to quality of life suffered during and between attacks, including interference with activities of daily living and heightened emotional distress.2 The frequency of attacks in patients can also vary over the course of their life, for example, during puberty or pregnancy or during stressful periods of life such as exams.1
Patients can experience symptoms in a range of locations on the body:7,8,9
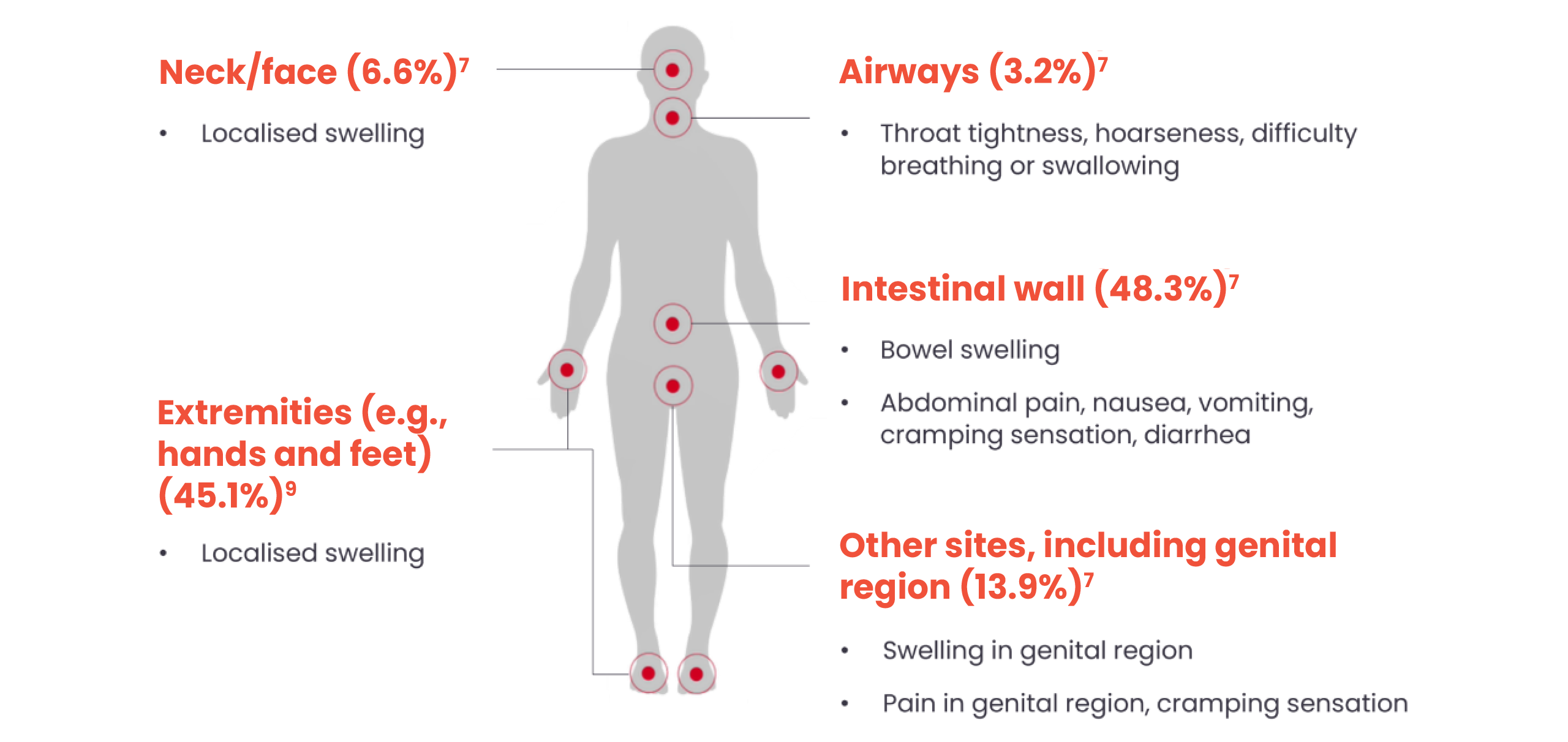
7The percentages indicate the number of HAE attacks affecting the body system in a global registry study (N=1297 patients; N=8885 attacks).
9Percentage based on 131,110 attacks in 209 patients in an analysis of outpatients at a German Dermatology Centre
Swelling of the skin is one of the most common symptoms patients with HAE may present with, affecting almost all patients with HAE, and can occur anywhere on the body. Most commonly, swelling occurs on the patient’s face, hands, feet and genitals, and it usually spreads to disfigure the affected site.9,10
Image from HAEimages.com.
break
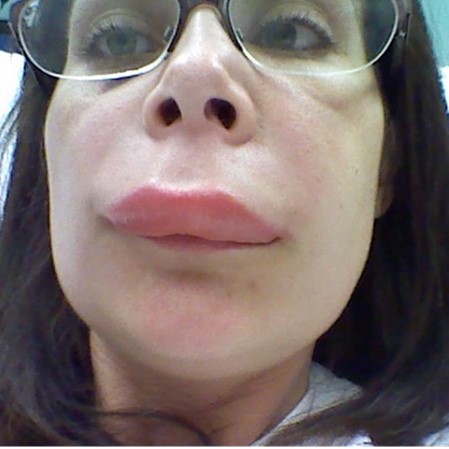
Image from HAEimages.com.
HAE attacks are often misdiagnosed as histamine-mediated/allergy-associated oedema, but it should be noted that attacks of HAE are nonpruritic and without wheals.1,8 Skin swellings are localised at a single site, affected by an extended oedema, and can grow and regress over 2–5 days.10
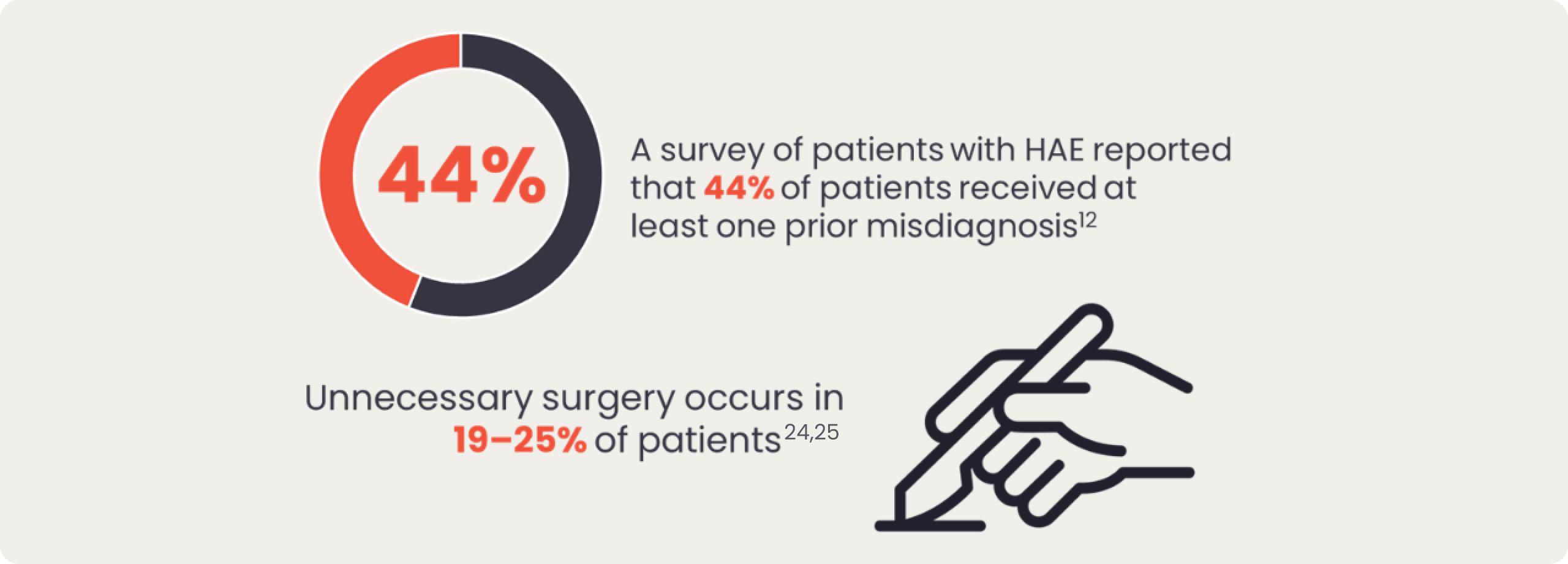
Patients with HAE may experience swelling of the gastrointestinal tract causing them to experience severe abdominal pain, nausea, vomiting and diarrhoea.2,11 In the case of severe abdominal pain caused by HAE swelling in the gastrointestinal tract, patients with HAE can be misdiagnosed with a food allergy/intolerance or appendicitis, which can result in unnecessary surgery.12 Approximately 20% of HAE patients may experience abdominal symptoms only, putting them at a particularly high risk for surgical interventions.13

Laryngeal oedema requires emergency attention, and can be life-threatening, particularly in patients who have not yet been diagnosed with HAE.14 Patients should be made aware of the signs of a laryngeal attack, which include:9

Prodromal symptoms act as signs of a potential attack and can vary between each person. Common indicating symptoms include:15


break
Erythema marginatum is a flat, non-pruritic, ring-shaped rash that most commonly affects the trunk in some patients prior to an HAE attack.16 It is more frequent in children, occurring in 42–58% of cases, and it should not be confused with urticaria.1
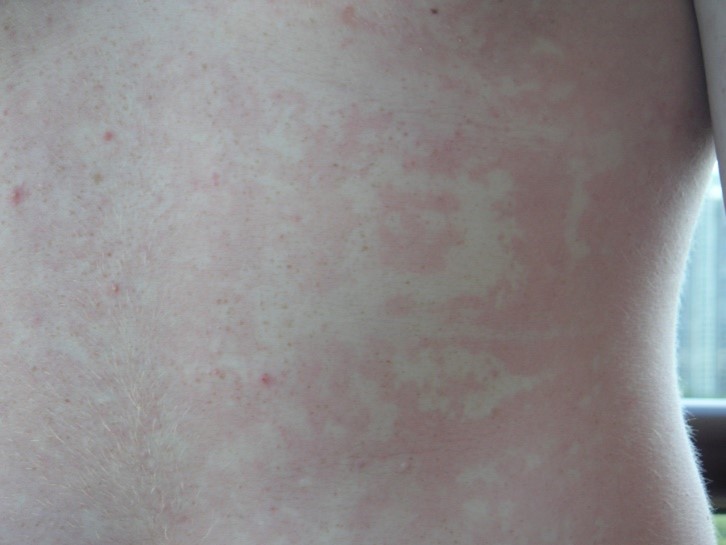
Erythema marginatum.
Image from HAEimages.com.
While most HAE attacks happen spontaneously and unexpectedly, there are various triggers that can cause an attack; however these may be specific for each patient with HAE.17 Some of the common triggers that may affect patients include:1,17-19
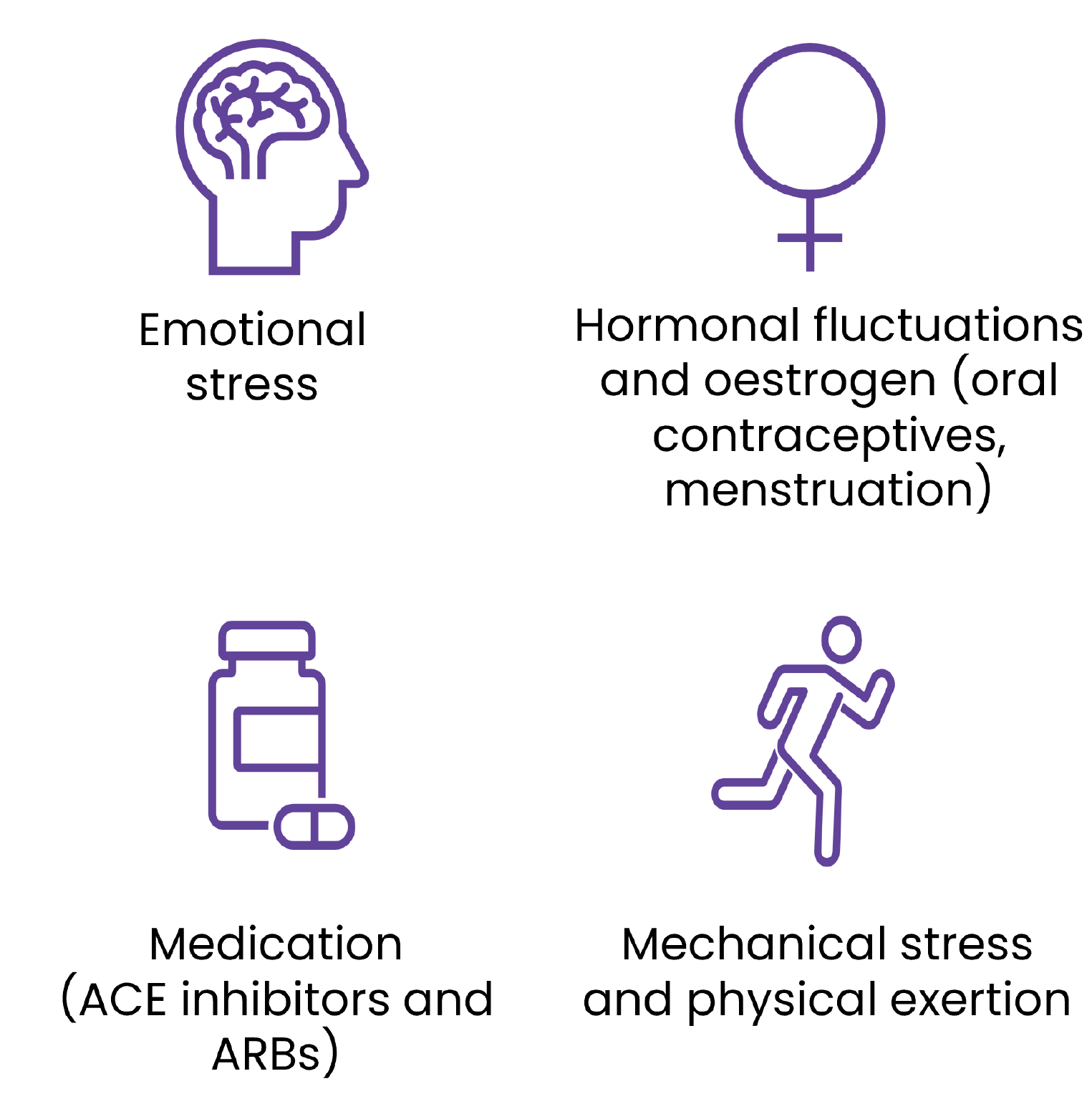
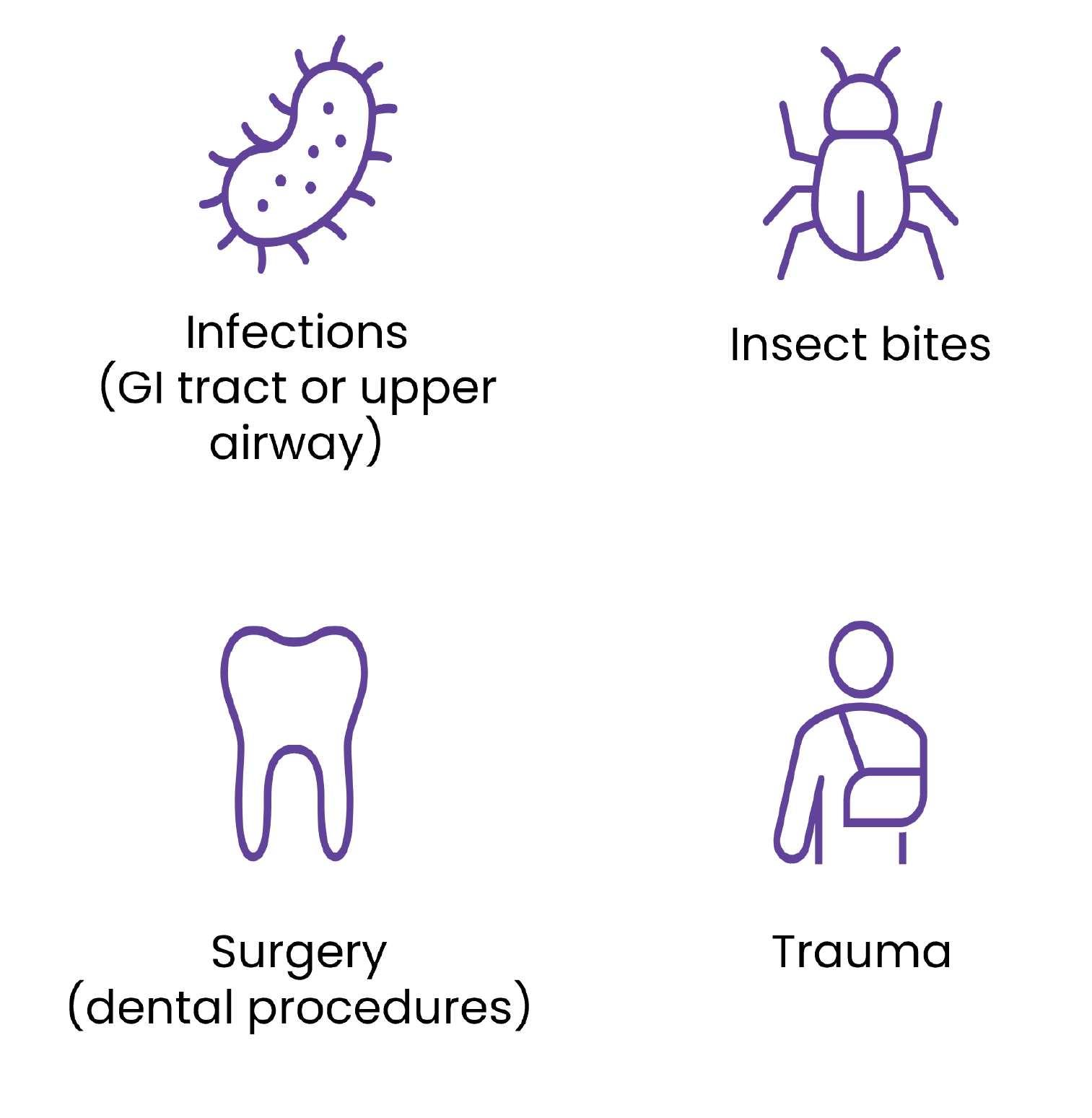
All patients with HAE should be educated about triggers that may induce attacks.
Some triggers can be prevented, for example, by avoiding the use of oestrogen containing oral contraceptives and ACE inhibitors. Vaccinations can also be given to prevent infections triggering attack, which is particularly common in children. Additionally, good dental care can reduce extractions, need for aggressive dental procedures and prevent acute or chronic intraoral inflammation.
Some triggers are an unavoidable part of a patient’s life, such as strenuous physical activities involving mechanical trauma and stress, so it is important that restriction of suspected triggers is individualised.
Excessive avoidance of suspected triggers should not be encouraged to avoid limiting the patient's normal life.1

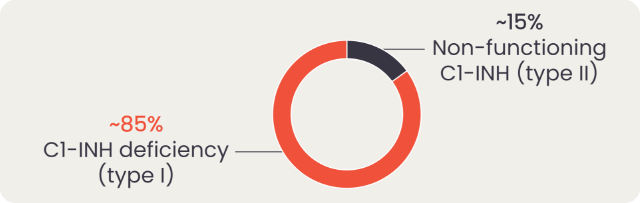
In most cases, HAE is caused by deficiency (type I) or dysfunction (type II) of C1-INH, that results in uncontrolled activation of the contact system and release of bradykinin.3
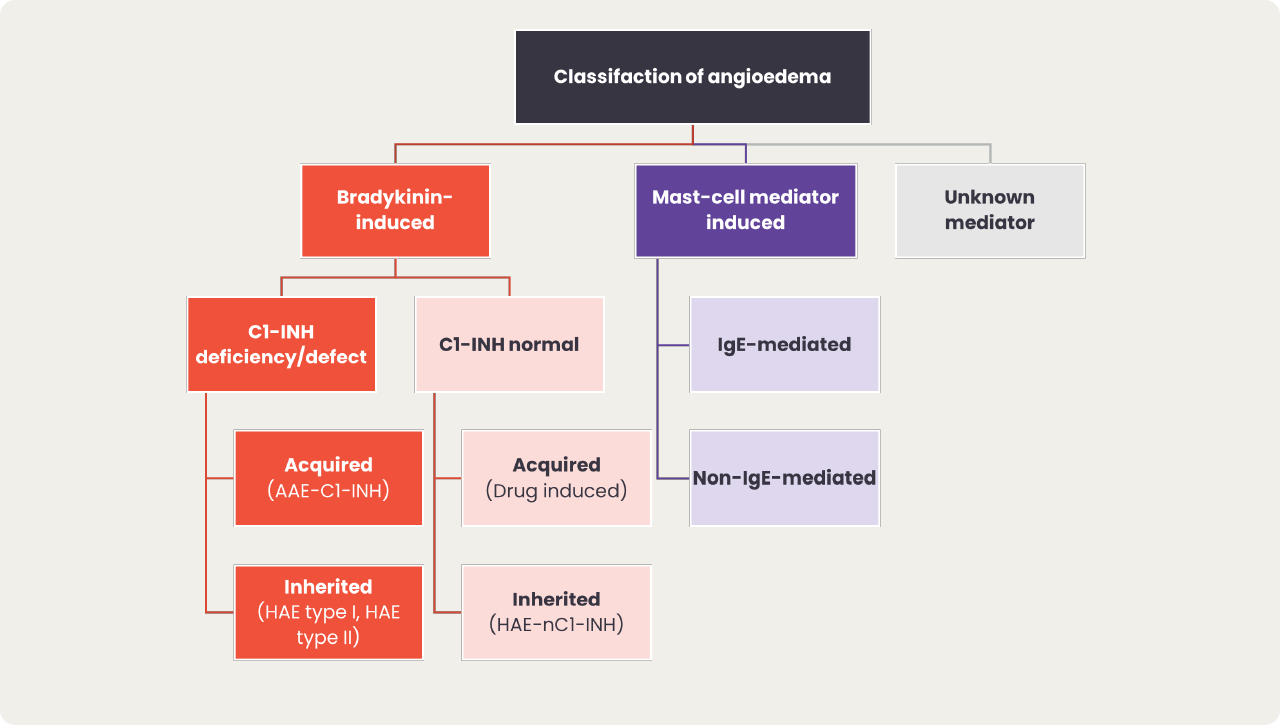
However, HAE can also be diagnosed in patients with normal C1-INH due to mutations in other genes. People diagnosed with HAE with normal C1-INH (HAE-nC1-INH) experience largely similar clinical symptoms as those with type I or II HAE.1
HAE-nC1-INH is classified by the gene in which the mutation occurs.
Currently there are 7 known HAE-nC1-INH types classified by the gene in which the mutation occurs;20,21
The most common type of HAE-nC1-INH is HAE-FXII, which has been reported in 20-25% of patients with HAE-nC1-INH.22 The mediator of angioedema varies between types of HAE-nC1-INH, and in some cases is still unknown.1,23
Diagnosis of HAE requires differentiation from mast cell-mediated angioedema and other forms of bradykinin-mediated angioedema to ensure effective treatment.1
Angioedema can be broadly split into bradykinin-mediated and mast cell-mediated, with the exception of idiopathic angioedema for which the mediator is unknown.1 Within bradykinin-mediated angioedema, we can further differentiate between C1-INH deficiency angioedema and angioedema with normal C1-INH, which can either be acquired or hereditary.
Due to the rarity of the disease, patients may have been previously misdiagnosed with more common conditions such as allergy-related angioedema, and they may have experienced long diagnostic delays.12 In the case of severe abdominal pain caused by HAE swelling in the gastrointestinal tract, patients with HAE can be misdiagnosed with a food allergy/intolerance or appendicitis which can result in unnecessary surgery.12
Maurer M, et al., Allergy. 20221
Click here for a detailed table summarising the key differences between angioedema types
Early and correct diagnosis of HAE is key, especially due to the potential for laryngeal swellings, which pose a risk of asphyxiation.14
When should HAE be suspected?1
After clinical suspicion has been raised, a simple blood test can be used to diagnose HAE by assessing:1

If tests confirm a diagnosis of HAE, it is recommended that the whole family also be tested, due to the genetic nature of the disease. The earlier HAE can be diagnosed, the quicker a suitable management plan can be implemented for the patient.1